V 1.0 The New (Old) Genetics
Abstract
The field of Genetics started flourishing after the rediscovery of the Mendelian laws of inheritance at the beginning of the 20th century. These laws are based on a discrete classification of phenotypes and their causative genes. Such a Mendelian way of thinking forms the foundation of modern molecular biology, with its experimental paradigm that a gene function is inferred from the knock-out of the gene. However, most phenotypes are not discrete. Human height, for example, is a continuous phenotype and height measures approximate a Gaussian distribution. The statistical foundation for the genetics of human height was worked out by GALTON at the end of the 19th century. He established the basis of quantitative genetics, a field that has driven the agricultural and breeding programs in the past century. It is not until very recently that the technical developments behind the human genome project have paved the way to reconcile the two contrasting ways of genetic thinking – Mendelian genetics and statistical genetics – through genome-wide analyses. It has now become clear that most phenotypes are rarely determined by single Mendelian genes, but instead, many genes contribute to their determination and variation. It has even been suggested in the omnigenic model that all genes that are expressed at the appropriate time contribute to any given phenotype. These insights are stimulating a major rethinking of how the linear genetic information laid down in the deoxyribonucleic acid (DNA) is converted into the threedimensional structure of an individual. The new conceptual and experimental paradigms have already revolutionized animal and plant breeding. In the field of human genetics, the realization that common diseases also have a polygenic basis is raising new challenges for treatment. And finally, in basic sciences like molecular and evolutionary biology, researchers are starting to revisit traditional, but oversimplified concepts on how genes act and how evolutionary adaptation works.
General Lay Summary
When one thinks of genetics, the first thing that comes to mind is Mendel. Mendelʼs laws are taught at an early age in school and for many school leavers they remain the only contact with genetics. Yellow and green peas are used to show how traits are inherited, ho w mixing and splitting can occur. However, while such categorial thinking has fueled the development of modern molecular genetics, it does not reflect the fact that neither the common phenotypes, nor their inheritance, can be described in these terms. Instead, the rules of quantitative genetics apply to most visible manifestations of organisms. The principles of quantitative geneticshave been worked out by a contemporary of Mendel – Francis Galton. However, they have led for a long time a shadow existence,only familiar to animal and plant breeders. Among quantitative geneticists, statistics prevail. There are no categorical distinctions, such as the green and yellow peas, but only continuous distributions, such as body size.The theoretical concepts for uniting Mendelʼs and Galtonʼs genetics had already been worked out by Ronald Fisher in the 1920s. He proposed two models of how the genetics of quantitative traits could be imagined. The first said that they were created bycombining the variants of a few genes, the second assumed the combined action of a great many genes, each with very small effects. The first model is close to Mendel’s genetics and was therefore long favored. The second model was rather dismissed as a mathematical ideal, but since a few years, the new data coming from genomic analyses indicate that the second model reflects reality much better than the first. With the tools of genomics, it is now possible to determine for each individual gene in the genome what proportion it makes up of a continuous phenotype, for example the body size. One can then ask how much of the total height is due to each of the genetic variants. To obtain such data, several hundred thousand individuals with millions of variants had to be screened. The results showed Fisher’s second model is much more likely, i.e. the combined action of many variants with small effects each. Some geneticists now even assume that ultimately all genes in a genome contribute to each phenotype in varying proportions, the so-called “omnigenic model”. While quantitative genetics teaches that there are no genetic categories, our thinking is still mainly shaped by categories. For example, we understand well how the categories “male” and “female” are determined by the distribution of the X and Y chromo-somes according to Mendel’s rules. However, quantitative genetics applies to the expression of the resulting sexual characteristicsand sexual behavior. There is no longer just the category male or female, but distributions and overlaps of characters. If our schools had always taught quantitative genetics, this would have prevented many misunderstandings of genetics and inheritance.
Generelle Zusammenfassung
Wenn man an Genetik denkt, kommt einem als Erstes MENDEL in den Sinn. MENDELS Gesetze werden schon früh in der Schule gelehrt, und für viele Schulabgänger bleiben sie der einzige Kontakt mit der Genetik. Gelbe und grüne Erbsen werden verwendet, um zu zeigen, wie Merkmale vererbt werden, wie Vermischung und Aufspaltung einzelner Merkmale auftreten können. Doch ob-wohl ein solches kategoriales Denken die Entwicklung der modernen Molekulargenetik beflügelt hat, spiegelt es nicht die Tatsache wider, dass weder die Körperformen und Verhaltensformen, noch deren Vererbung mit diesen Begriffen beschrieben werden kön-nen. Stattdessen gelten für die meisten sichtbaren und messbaren Ausprägungen von Organismen die Regeln der quantitativen Genetik. Die Prinzipien der quantitativen Genetik wurden von einem Zeitgenossen von MENDEL – Francis GALTON – ausgearbeitet. Sie haben jedoch lange Zeit ein Schattendasein geführt und waren meist nur Tier- und Pflanzenzüchtern bekannt. In der quanti-tativen Genetik herrscht die Statistik vor. Es gibt keine kategorialen Unterscheidungen, wie grüne und gelbe Erbsen, sondern nur kontinuierliche Verteilungen, wie z. B. die Körpergröße.Die theoretischen Konzepte zur Vereinigung von MENDELS und GALTONS Genetik wurden bereits in den 1920er Jahren von Ronald FISHER ausgearbeitet. Er schlug zwei Modelle vor, wie man sich die Vererbung von quantitativen Merkmalen vorstellen könnte. Das erste besagte, dass sie durch die Kombination der Varianten einiger weniger wichtiger Gene möglich sein könnte, das zweite ging von der kombinierten Wirkung sehr vieler Gene aus, jedes für sich mit jeweils nur sehr geringen Auswirkungen auf das gemessene Merkmal. Das erste Modell steht der Mendelschen Genetik nahe und wurde deshalb lange Zeit favorisiert. Das zweite Modell wurde eher als mathematisches Ideal abgetan, aber seit einigen Jahren deuten die neuen Daten aus der Genomanalyse darauf hin, dass das zweite Modell die Realität viel besser widerspiegelt als das erste. Mit den Werkzeugen der Genomik ist es nun möglich, für jedes einzelne Gen im Genom zu bestimmen, welchen Anteil es an einem kontinuierlichen Merkmal hat, z. B. der Körpergröße. Man kann dann fragen, wie viel von der Gesamtgröße auf jede der genetischen Varianten zurückzuführen ist. Um solche Daten zu erhalten, mussten mehrere hunderttausend Individuen mit Millionen von Varianten durchmustert werden. Die Ergebnisse zeigten, dass das zweite Modell von FISHER viel wahrscheinlicher ist, also die kombinierte Wirkung vieler Varianten mit jeweils nur geringen Auswirkungen jeder einzelnen. Einige Genetiker gehen heute sogar davon aus, dass letztlich alle Gene in einem Genom in unterschiedlichen Anteilen zu jedem Merkmal beitragen, das sogenannte „omnigenic model“. Während die quantitative Genetik lehrt, dass es keine genetischen Kategorien gibt, ist unser Denken immer noch hauptsächlich von Kategorien geprägt. Wir verstehen z. B. gut, wie die Kategorien „männlich“ und „weiblich“ durch die Verteilung der X- und Y-Chromosomen nach den Mendelschen Regeln bestimmt werden. Quantitative Genetik gilt jedoch für die Ausprägung der resultierenden Geschlechtsmerkmale und des Sexualverhaltens. Es gibt nicht mehr nur die Kategorie männlich oder weiblich, sondern Verteilungen und Überschneidungen von Merkmalen. Hätten unsere Schulen schon immer quantitative Genetik gelehrt, hätte dies viele Missverständnisse der Genetik und Vererbung vermeiden können.
Keywords
Background
The foundations of modern genetics were independently laid down in the second half of the 19th century by Gregor MENDEL (1866) and Francis GALTON (1889). While MENDEL emphasized the independent combination of discrete characters, such as green versus yellow peas, GALTON focused on the statistical principles that describe continuous phenotypes, such as human height. This is not to say that Mendel was not aware that the characters he chose were not as discrete as he would have wished. But “seeing” of simple patterns at the expense of complexity was one of his major achievements, largely as a perceived necessary strategy to penetrate the complexity (WEEDEN 2016). Up until today this distinction of discrete versus continuous characters endures in reflecting the fundamental dichotomy in looking at mechanisms of inheritance. In fact, it is rather comparable to the particle-wave duality of quantum mechanics. EIN- STEINʼS quote on this duality could almost seamlessly be applied when exchanging the term “light” with “genetics”:
It seems as though we must use sometimes the one theory and sometimes the other, while at times we may use either. We are faced with a new kind of difficulty. We have two contradictory pictures of reality; separately neither of them fully explains the phenomena of light, but together they do.” (EINSTEIN and INFELD 1938, p. 262.)
But in contrast to quantum physics, in biology the basic theory for resolving this duality was worked out in the early 20th century, most systematically by Ronald FISHER. He formulated what is nowadays called the infinitesimal model of inheritance:
“The simplest hypothesis […] is that such features as stature are determined by a large number of Mendelian factors, and that the large variance among children of the same parents is due to the segregation of those factors in respect to which the parents are hete- rozygous.” (FISHER 1918, p. 400.)
However, experimental proof of FISHERʼS “large number of Mendelian factors” was lacking for a long time and is only now beginning to emerge. In fact, Mendelian genetics – understood as one gene, one di- screte phenotype – has prevailed in text books and public understanding. This is even embodied in con- ventions of gene naming, where genes are often named according to their mutant phenotype. Since discrete phenotypes can be measured accurately, the experimental results are more straightforward and easier to communicate. This results in a perceived value in suggesting a simple mechanistic understand- ing of phenotypic variation. Even in physics we still prefer mechanical over quantum laws, although both are required to explain the real world.
MENDEL’S genetics has laid down the foundations for molecular genetics and is still dictating its ex- perimental paradigms. GALTON’S genetics, on the other hand, laid down the foundations of quantitative genetics, correlative statistics and biometry (further worked out by Karl PEARSON and Raphael WELDON, who teamed up with GALTON in the early 1900s). This then became the basis for the progress in agricultural plant and animal breeding in the 20th century. A reunion of these rather separate develop- ments – molecular and quantitative genetics – has only started with the genomic revolution in the 21st century, especially with the developments in genome-wide association studies (GWAS) that allow the simultaneous interrogation of all genes in the genome for their effect on a given phenotype. This is already revolutionizing breeding experiments and the understanding of complex diseases. We are still only in the beginning of these developments, where new concepts and insights are being turned out at a high rate. We are entering a phase where the long-standing unsolved question of the relationship between the genotype and the phenotype is receiving new impulses and fresh experimental paradigms are starting to emerge. This will have major impacts on further disciplines. In evolutionary biology it will help to understand the mechanisms of genetic adaptation. In developmental biology it will help to understand how the linear information from the DNA gets converted into the three-dimensional Gestalt of an individual. And we will finally have a better understanding of the role of the environment versus genetic determinism in the formation of the Individuum. There will also be major consequences for our under- standing of complex diseases, including a rethinking of strategies for developing new pharmaceuticals.
Galton’s Legacy
“[...] to show the large part that is always played by chance in the course of hereditary transmission, and to establish the importance of an intelligent use of the laws of chance [...] in expressing the conditions under which heredity acts.” (GALTON 1889, p. 17.)
GALTON was interested in understanding the mechanisms of heredity throughout his scientific life. While focusing a lot on inheritance patterns in humans, he also did breeding experiments with sweet-peas and moths. Especially with the latter, he could also have discovered MENDEL’S rules, but his focus was on continuous phenotypes, rather than discrete ones. He realized that continuous phenotypes can only be described with a statistical treatment of the group of relatives from the preceding generations, while MENDEL had focused on describing phenotypic categories without including a statistical treatment. The statistical techniques and principles that GALTON developed and discovered include correlation, the im- portance of the median, the concept of standard deviations, the regression to the mean, the use of ques- tionnaires, and, based on twin studies, the role of the environment for expressing the phenotype.
His focus on human traits and breeding led him inevitably to the question whether breeding humans towards a better phenotype would make sense. He coined the terms “dysgenics” and “eugenics” for this approach, evidently without anticipating the horrible implications that it will have in later decades. Rather, he took a very scientific attitude towards this question and clearly cautioned:
“[…] but its details must first be worked out sedulously in the study. Overzeal leading to hasty action would do harm, by holding out expectations of a near golden age, which will certainly be falsified and cause the science to be discredited.” (GALTON 1904, p. 6.)
This was rather visionary, given that politically motivated eugenics did indeed lead to the thorough dis-creditation of eugenics.
GALTONʼS most important book on the principles of heredity is Natural Inheritance (GALTON 1889). It is an extremely scholarly book, solidly based on data and innovative ways of analysis (and eugenics plays no role in it). It remains very readable, being full of explanations and thoughts that led to his conclusions and largely remain valid until today. It would be fair to say that in its implications it should be con- sidered as being comparable with his half-cousins’ book on the Origin of Species (DARWIN 1859) and it would deserve to be equally hailed. But it was apparently received with much more skepticism (STANLEY 1889).
One of GALTONʼS great ideas to obtain data for statistical analysis was the public offering of prizes for sending him personal data. He published an advertisement in newspapers starting with “Mr. Francis GALTON offers 500£ in prizes to those British Subjects resident in the United Kingdom who shall furnish him before: May 15, 1884, with the best Extracts from their own Family Records.” (GALTON 1889, p. 72), followed with detailed descriptions of what he wanted. And, not much different from today, people were very willing to provide their personal data when a reward is promised (e.g. use of a “free” internet service). GALTON was at that time mostly interested in the inheritance of human height, since this is a continuous trait that can be objectively measured. He received useable datasets from 150 extended fami- lies and given that these families were much larger in these times, these data remain valuable until today. GALTON used these data (together with other data from eye color, as well as his experiments on peas and moths) to work out key principles of quantitative genetics. He showed that the height measurements had a statistically normal – or Gaussian – distribution (a hallmark of all quantitative phenotype data), that there was a sex-specific effect that he could correct with a single factor and that the midpoint average between the mother’s and the father’s height correlated linearly with the height of the offspring. But he noted also that the offspring of tall parents tended to be somewhat smaller than expected from a pure lin- ear correlation and the offspring from short parents tended to be somewhat larger. He called this effect “regression to mediocrity” (nowadays: regression to the mean) and it is by now well understood that this is a general effect of statistical sampling (BARNETT et al. 2005). To GALTON this suggested that many independent “particles” must be involved in height determination and that these were passed on over the generations. One of his key conclusions was therefore:
“We appear, then, to be severally built up out of a host of minute particles of whose nature we know nothing, anyone of which may be derived from any one progenitor, but which are usually transmitted in aggregates, considerable groups being derived from the same progenitor.” (GALTON 1889, p. 9.)
He provides there an almost modern description of a polygenic inheritance with chromosomal linkage.
Although GALTONʼS work on heredity was ground breaking and has led to many further developments in statistics and animal and plant breeding, its true value for understanding core principles of genetics has been largely overlooked for more than a century. Intriguingly, the imminent resurrection of these principles in the genomics age has come again from human height studies. The most extensive GWAS that have been done so far, for any system, are on human height, and they have shown that the principles of Mendelian genetics that have dominated the last century of genetics research need to be complemented by statistical genetics. Gene functions have both a particulate, as well as a statistical nature and both need to be considered for a full understanding of genetic architectures and their products, the living organism.
Continuous versus Discrete Thinking Regarding Human Traits
One of the hallmarks of continuous phenotypes is that their measurement values can often be approximated by a Gaussian distribution (sometimes informally also called a bell curve, HERNSTEIN and MURRAY 1994). GAUSS had derived this distribution from error theory to identify the real value from a set of measurements with errors. As discussed above, this made GALTON right from the beginning believe that there must be a statistical sampling effect among many “particles” derived from progenitors to explain this observation. But initially, this turned out to be in contrast to MENDELʼS laws when they were rediscovered at the beginning of the 20th century. These laws state that discrete genotype classes are formed through the combination of alleles of a single gene. This should usually result in discrete phenotype categories, rather than a continuous Gaussian distribution. In fact, this is the fundamental clash between what is taught in school about Mendelian genetics and what the real-life experience of inheritance is. The phenotype categories that lead to the Mendelian rules are very useful abstract concepts, but the actual phenotypes that one encounters almost never fall into simple categories. This led to many misunderstandings, especially with respect to the common thinking that “a gene” would determine a given trait. But in human genetics, there are practically no cases of such simple scenarios for normal phenotypes.
The typical school examples for teaching Mendelian rules include eye color determination in humans. It is still often portrayed as being determined by a single gene with two alleles, whereby brown eyes are said to be dominant to blue eyes. Under this model, parents with blue eyes could not have a child with brown eyes. Although this is indeed uncommon, parents with blue eyes can have children with brown eyes since there are at least 16 genes responsible for eye color, with complex interactions among them that can produce different color outcomes (WHITE and RABAGO-SMITH 2011).
The other main school example is the genetics of sex determination through sex chromosomes. This is indeed nominally very simple, since the combinations of the sex chromosomes X and Y yield (at least in most mammals) a female in the XX combination and a male in the XY combination i.e. two categories (Fig. 1A). But “male” and “female” are not distinct categories at the level of their adult phenotypes. Just the opposite, secondary sexually determined characters are variable between individuals and when they are quantitatively measured, they can fall into continuous, but overlapping Gaussian distributions (MANEY 2016), as it is, for example, the case for body height (Fig. 1B).
Hence, it is clear that other genetic principles are required to explain the determination of secondary sexual characters that emerge after the initial sex determination step is completed. Interestingly, attempts have nonetheless been made to draw a picture of a typical male and female, e.g. in the case of the engrav- ings for the plaque on the Pioneer 10 and 11 spacecrafts which left the solar system. The plaques are meant to give aliens an idea where the spacecrafts came from and how we look like when they find the probe in outer space (Fig. 1). Hence, we naturally entertain the idea of the existence of a typical male and female phenotype, but reality is different. Why was there no attempt to symbolize the variation of human phenotypes? Would aliens be confused when they find variation, rather than types? Or is the as- sumption of variation around types universal, i.e. aliens would themselves be variable?
The categorization into typical male and female phenotypes seems a natural social game, with vivid discussion on the role of genetics versus environment in shaping the phenotype. This ranges from sexual behavior to the morphology and function of the brain. For sexual behavior genome wide analyses show that there is no single allele that has a major influence on this behavior. Instead, sexual orientation is based on a combination of polygenic effects and environmental influences (GANNA et al. 2019). The corresponding behavioral phenotypes are therefore bound to overlap, similar as height. The suggestion of sex differences in the brain has also drawn much attention, starting from physiological and morphological observations to developmental and hormone regulation considerations (MANEY 2016). Given that each of these characters is most likely itself polygenically determined, it is not surprising that the relation- ships must be complex (JOEL et al. 2020). In a systematic analysis of whether gender differences are categorical or dimensional, REIS and CAROTHERS (2014, p. 19) concluded that “[...] for the psychological constructs that we examined, there is little support for believing that sex differences are anything more than individual differences that vary in magnitude from one attribute to another.”
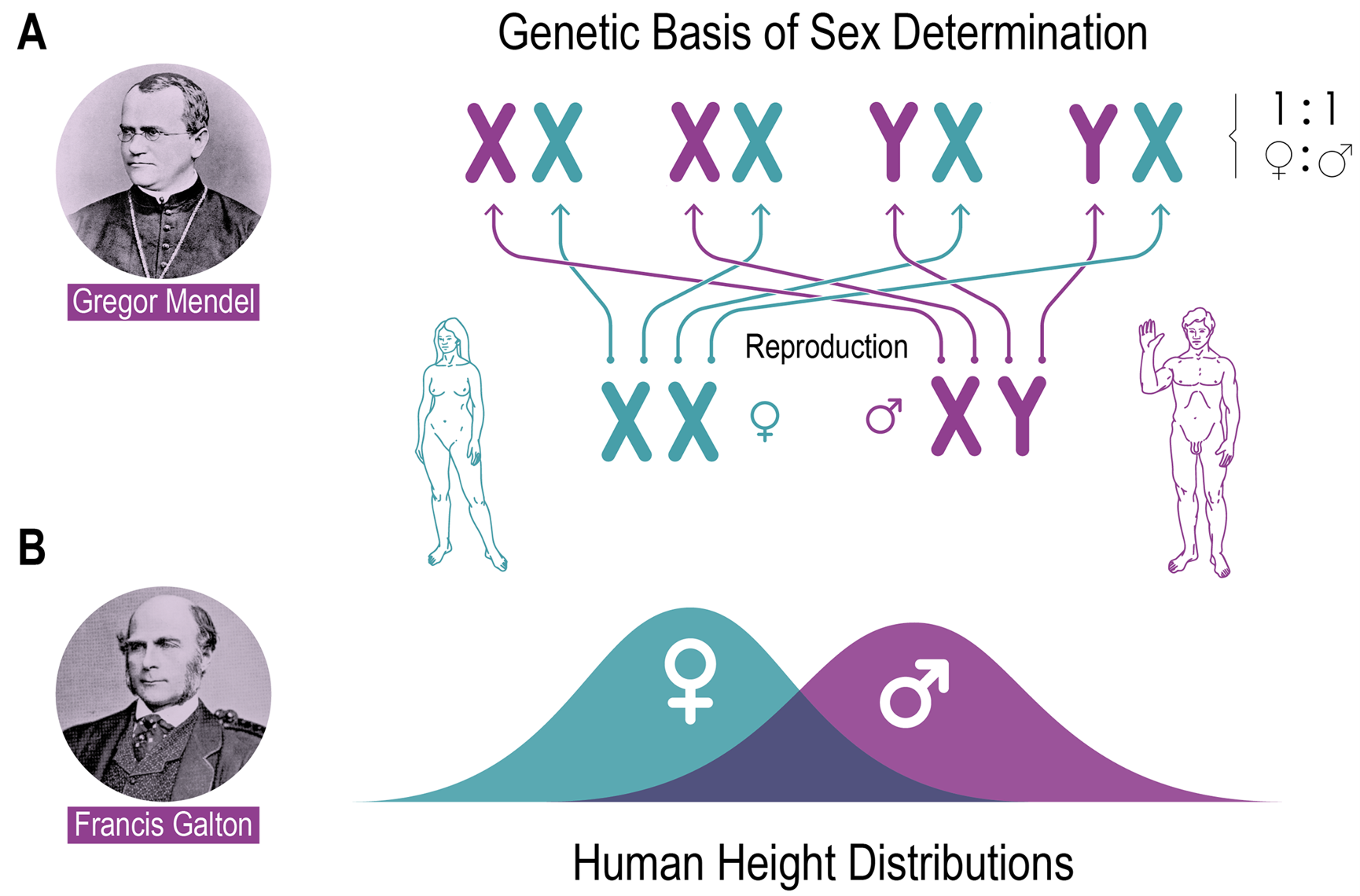
Fig. 1 Comparison of the two genetic concepts in case of sex determination. Panel A shows the Mendelian concept with two discrete sex chromosomes, which combine independently to form the next generation and create two discrete categories of sex. Mixing the gametes according to the Mendelian laws in the next generation recreates the sexes with a 50 % probability each and thus keeps a constant sex ratio with half males, half females in the population. Panel B relates to what follows after this initial step, namely the formation of the actual phenotype according to the principles discovered by GALTON. This results in continuous traits that form Gaussian distributions, here shown as the height distributions for females and males. The variance of these traits is gene- rated as a mixture between genetic and environmental factors, which can lead to different averages in the sexes, but with substantial overlap. The male and female figures are the symbolic categorial figures that were used in the engravings of the Pioneer 10 and 11 spacecrafts and reflect the typological thinking about sex differences (see text).
A particularly problematic form of discrete thinking has emerged in the discussion around the inheritance of intelligence. The measurement of “intelligence” via intelligence quotient (IQ) tests yields continuous values and any discussion of its heredity should therefore be fully in the realms of GALTON’S genetics. But some authors have assigned average IQ values to subpopulations and implicitly treat these as discrete characters for these populations. When this is combined with poorly substantiated claims of differences in average fertility for these population groups, they end up with scenarios of predicted substitutions of populations. But such discrete thinking is alien to quantitative genetics and this line of arguments is there- fore wrong on many accounts, even independent of the question whether IQ can be objectively measured. First of all, even if only parents from one end of the distribution would have children, one would still re- create almost the same variance in IQ distribution as before (see Box 1, part B). Only the population average could slightly change, and even this would not happen as long as there is at least some population mixing. Further, any prediction on changes of averages depends on the measure of heritability and this is in itself not a constant factor, but depends on the condition under which it is measured (Box 1). Hence, it would dramatically help in such a discussion if the principles of quantitative genetics would be taught in school alongside the principles of Mendelian genetics.

Oligogenic versus Polygenic Concepts
As discussed above, the principle solution to bridge the gap between categorical and continuous phenotypes has been worked out by FISHER (1918, 1930). It starts with the realization that if more than one gene contributes to a phenotype, then one gets even under strict Mendelian rules an increasing number of genotype classes. Under a random assortment model, these classes can result in a phenotypic distribution that approximates a Gaussian distribution. For example, if two genes with three alleles each con- tribute to a phenotype, then the Mendelian mixing of these yields already 9 phenotypic classes (Fig. 2). Hence, from a Mendelian point of view, the problem of continuous traits could simply be solved by considering a mix of a handful of genes, the so-called “oligogenic” model. In this vein, THODAY and THOMPSON (1976, p. 335) conducted a study starting with the premise:
“Phenotypic distributions indistinguishable from normal distributions are often interpreted as showing the segregation of many genes. Such characters may then be passed over by physiologists and developmental biologists as too complex or otherwise unsuit- able for developmental studies.”
to show that the great majority of observed variance could potentially be explained by few loci, which would make it much more accessible to mechanistic studies. This reflects a sentiment of convenience for interpreting genetics that was upheld until recently. But FISHER had proposed a polygenic model that implied the contribution of very many genes, each with very small effects (often called “infinitesimal model” to differentiate it from the oligogenic model). This reflected GALTON’S view of very many “particles” that should randomly combine to form a Gaussian distribution (Fig. 2). Unfortunately, for a long time it was almost impossible to experimentally distinguish between these alternatives.
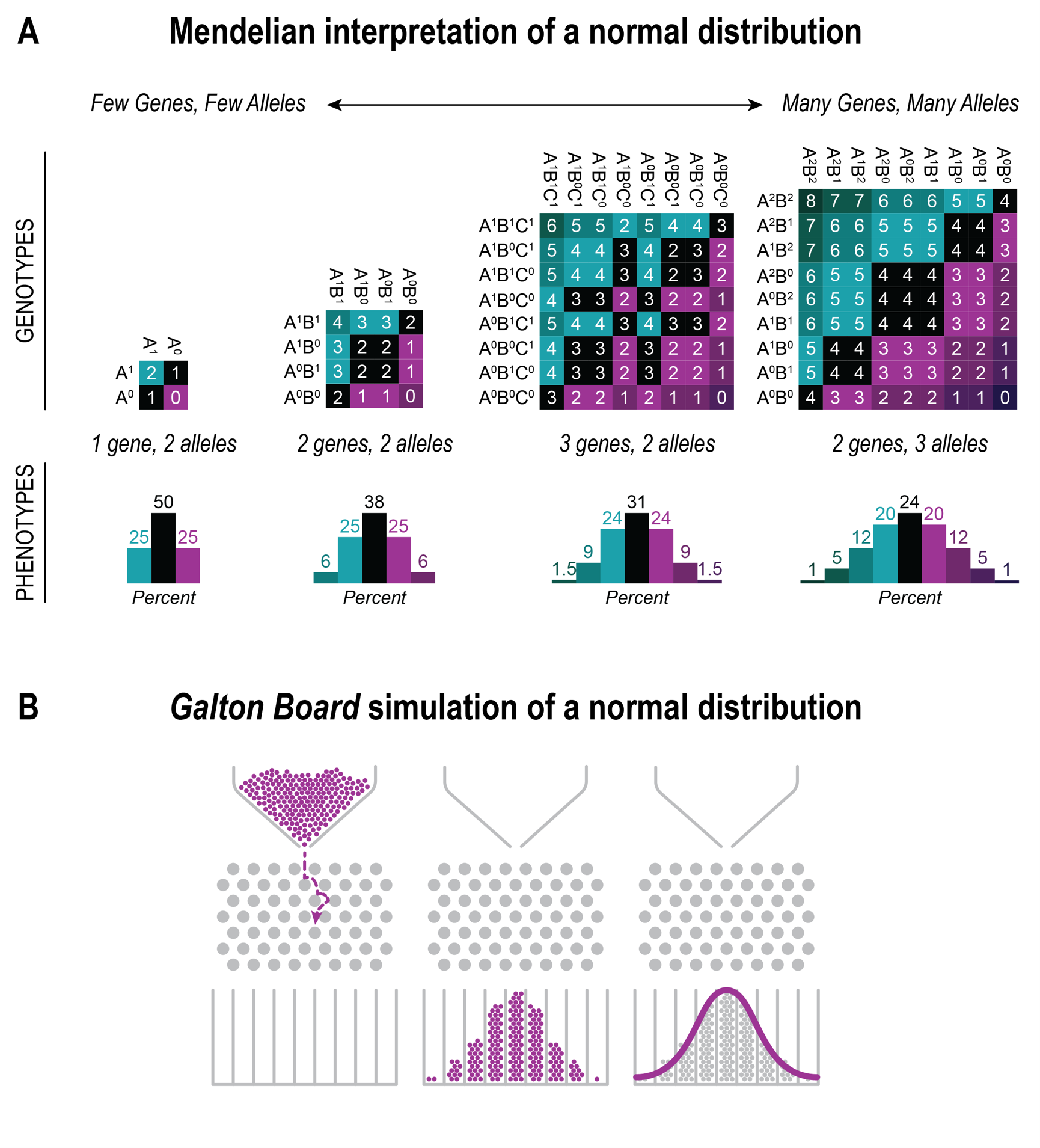
Fig. 2 Comparison of concepts on how the Gaussian distribution of continuous phenotypic measures can be explained. (A) The
Mendelian based interpretation under the assumption of the combination of a small number of genes or alleles. (B) The random
simulation scheme devised by GALTON. GALTON first described this apparatus in his book (GALTON 1889) (now often called
Galton board) to demonstrate basic principles of statistics.
But there is also a further complication. MENDEL (and his followers) had shown that variants of a gene
(alleles) can be dominant over other variants, i.e. in the presence of one copy of a dominant variant, only
this has a phenotypic effect. Further, different genes can be epistatic over each other, meaning that the
expression of one gene is required to allow another one to have an effect on the phenotype. This is for
example the case in a cascade of enzymes that create a color variant. In a polygenic model, both dom-
inance and epistasis lead to many additional possible combinations of phenotypic effects that need to be
considered. Although these effects can be incorporated into the infinitesimal model (BARTON et al. 2017),
this has the unfortunate consequence that a full statistical treatment becomes next to impossible in real
experiments. Hence, these statistics require the simplified assumption of all alleles and genes acting only
in an additive way. The statistical treatment of non-additive effects remains therefore one of the great
problems that await a resolution.
Although many refined statistical approaches were devised to interpret the available experimental
data in at least one way or the other (i.e. oligogenic or polygenic concepts), the matching of actual genes
with quantitative trait phenotypes was an almost unsolvable challenge for decades. Nevertheless, quan-
titative genetics flourished among breeders because even without knowing the identity or number of the
underlying genes, the breederʼs equation (Box 1) allowed them to predict the response to artificial se-
lection and therefore to improve their breeds. Hence, quantitative genetics became extremely important
in practical terms since it led to significant improvement of todayʼs plant and animal live stocks that yield
our food (Fig. 3).
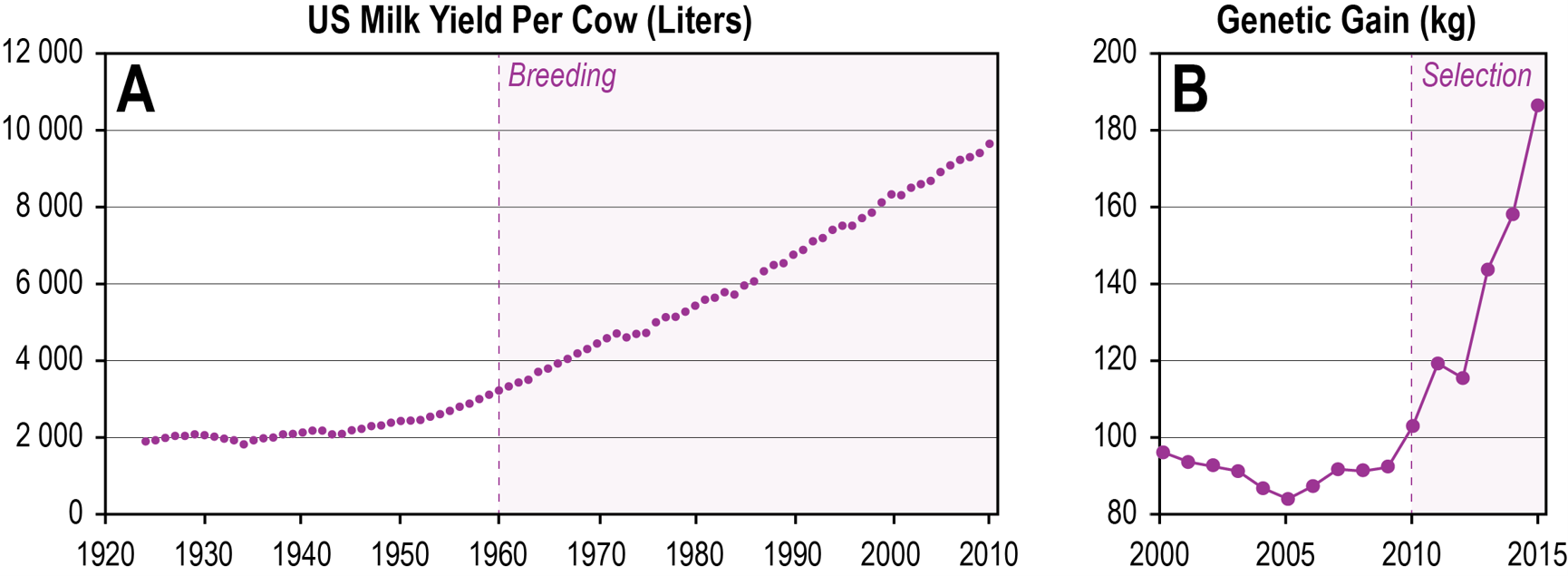
Fig. 3 Impact of quantitative genetics on milk production. (A) The chart represents a plot of liter milk produced per cow per year
in the US (Y-axis) in the past hundred years, with the gains due to systematic breeding starting in the 1960s (data from USDA
www.nass.usda.gov/Quick_Stats/Lite/). (B) The chart shows the impact of genomic selection on the yearly genetic gain in breeding value for milk yield (kg) (Y-axis), which has started around 2010 (data from GARCIA-RUIZ et al. 2016). Note that the relative gain had remained rather constant until then.
The Success of Mendelian Genetics
While quantitative geneticists struggled with finding experimental approaches to identify the genes behind the statistics, the Mendelian approach became extremely fruitful, especially after artificial mutagenesis and recombination mapping were discovered. MENDEL used naturally occurring variants of a gene (alleles) for his experiments, but by using artificial mutagenesis (initially either with chemicals or ionizing radiation, nowadays also with directed approaches, e.g. CRISPR/Cas mutagenesis), one could
create new variants that do not occur in nature but still serve to study the function of genes. The use of artificial mutagenesis resulted in a genetic paradigm that we may call “knockout genetics”. This paradigm states that the function of a gene is defined through its loss of function. In other words, one has to search for a mutant version of the gene which abolishes its function (a so-called “null allele”). Then, one can compare individuals that carry two copies of the null allele (i.e. homozygous mutants) with non-mutant individuals and infer the function(s) of the gene based on the differences between the two groups of individuals. While this approach has its pitfalls and limitations, it became hugely successful in the past decades given that it provided a practical answer to the question of gene function. This culminated in a research agenda that posited that one should be able to use systematic mutagenesis of all genes in the genome, combined with analyzing them one by one, to find every gene that contributes to a given phenotype(NOLAN et al. 2000). In Mendelian thinking, this is a perfectly valid assumption and there are endless examples where this has worked successfully to identify key genes in genetic pathways, up to the Noble prize for unraveling the genetic basis of early development in Drosophila (NUESSLEIN-VOLHARD and WIESCHAUS 1980). Intriguingly, if one would have adopted only GALTON’S genetics, one would not even have considered such an approach.
The principles of quantitative genetics are based on genes having naturally occurring functional variants or alleles. This in part explains the common mismatch between genes identified through homozygous knockout studies and genes identified with GWAS for the same trait – even when a gene (or its
absence) modifies a trait, it will only be identified in GWAS if it has naturally occurring functional variants that have been sampled.
The success of knockout genetics has led to a peculiar type of dogmatic thinking. It posits that thefunction of a gene has only been conclusively demonstrated when one has done a knockout, ideally combined with secondary artificial rescue with its wildtype variant. But this dogma is too simple, since it ignores what we know from quantitative genetics. In the polygenic model, none of these genes works in isolation, (very) many are expected to contribute to a given phenotype, implying also that single genes
can contribute to many phenotypes (this well-known phenomenon is called pleiotropy). In fact, even the knockout geneticists are perfectly aware that the phenotypic effect of a gene knockout depends on the genetic background of the individual (CHANDLER et al. 2013, SITTIG et al. 2016) which dictates an exper- imental paradigm that requires strictly controlled isogenic strains to obtain reproducible results. Hence, natural variation is excluded as much as possible from such studies.
Moreover, loss of the same gene in different species can have different phenotypic consequences, it can be lethal in some species and have almost no effect in other species (RANCATI et al. 2018). This implies that the function of a gene is part of a network and the evolution of this can change the core function of the gene. Even individuals in the same species can show very different responses to the loss of a gene.In humans, it is possible to detect cases where a healthy individual is mutant for a gene that would have severe medical consequences in most other individuals (NARASIMHAN et al. 2016).
Finally, while it is well known in quantitative genetics that environment plays a role in shaping the genetic effects on the phenotype, the Mendelian experimental paradigm tries to cut out the environmental effect as far as possible through standardizing the conditions under which one conducts an analysis. Unfortunately, there are still many misunderstandings, both with respect to experimental agendas, as well as to interpreting data when one takes only the Mendelian genetic paradigm into account.
Genome-Wide Association Studies (GWAS)
Genome-wide association studies (GWAS) have become the key to understanding polygenic inheritance. The experimental idea behind them is very simple, but the technical possibilities to run them at a meaningful scale have only recently emerged and are still being improved. The driving factor behind GWAS was the quest to understand the genetics of common genetic diseases in humans and to develop drugs against them.
In the simplest form of a GWAS one would have two groups of individuals from a natural population, one with a phenotype that one wants to map (e.g. a disease) the other a wildtype (or disease-free) group. Ideally one would then obtain the genome sequences from all individuals and compare them at every position to see whether certain genetic variants are more prevalent in one group versus the other. One of the main problems to achieve a truly genome-wide search is that full genome sequencing of properly sized cohorts (thousands to hundreds of thousands of individuals) is too expensive. The alternative has been to type all common variants in a population which are much fewer than all positions in the genome. This approach made the first human GWAS possible about 15 years ago (BURTON et al. 2007) and it remains the common standard until now. However, after the initial five years of accumulating data, it seemed that the promises to justify the investment were not met. In particular, the hope to find key genes for drug targeting of common human diseases fell apart and the whole approach started to be questioned.
VISSCHER et al. (2012) provide an account of the common critique at that time and respond to it. But regardless of what impac GWAS results have had on understanding complex genetic diseases, it is nonetheless clear that they have led to a revolution in genetics and biology.
Lessons from Human Height Studies
A natural trait that has changed our thinking in terms of single gene effects versus polygenic genetic architectures is human height. GALTON’S work had paved the way for its genetic analysis, but the question of which actual genes are involved in its regulation remained open. Physiologists had found in the 1950s a single hormone, somatotropin, that can control the body size of mammals in a concentration depended way. But how would such concentration differences come about in natural populations? It would require a multitude of alleles of the gene that should segregate in the populations in a way to create a continuous distribution. But this does not match with what we know about segregating alleles of a gene – most genes have only a few functional variants in an average population. In humans, height was an ideal model trait to develop techniques and procedures for large-scale genome-wide studies. The trait can be objectively measured in large cohorts, its heritability is very high, experimental analyses can be scaled and data can be easily combined from different studies. A first comprehensive study was published in 2010 and showed that variants in at least 180 loci contribute to human height (ALLEN et al. 2010). A later meta-analysis of several human height studies including data from more than 250,000 individuals was published in 2014 (WOOD et al. 2014). It provided unequivocal evidence that human height is indeed controlled by very many genes with many naturally segregating alleles. The authors identified at least 423 loci in multiple biochemical pathways, with thousands of causal variants. They concluded:
“Our results indicate a genetic architecture for human height that is characterized by a very large but finite number (thousands) of
causal variants.” (WOOD et al. 2014, p. 1173)
But although this showed very clearly a highly polygenic architecture, even this conclusion was still somewhat premature. They struggled with a problem that has turned up in practically all GWAS, namely that the sum of genetic effects calculated from the identified variants did not fully explain the observed heritability of the phenotype. The explanation for such a pattern was left open to speculation. In the meantime it has become clear that this “missing heritability” problem (MANOLIO et al. 2009) is mostly due to self-constraints in the experimental strategy and the statistical analysis, and although there have been many viewpoints of how the missing heritability in GWAS could be explained (EICHLER et al. 2010) it is now assumed that it has mostly two major reasons.
The first is the statistical significance cutoff for calling an allele significantly associated with a pheno- type (YANG et al. 2010). These cutoffs are chosen under the assumption that most of the association signals are noise and only a few can be real – an assumption that is grounded in the oligogenic view. Hence, one uses extremely strict statistical correction procedures to minimize the perceived chance of detecting “wrong” or only weak associations. But the polygenic view assumes that the phenotypes are caused by very many real, albeit small, effects. In fact, it can be shown that by not imposing a cutoff and simply adding up all association effects, one can much better explain the full heritability (YANG et al. 2011, VISSCHER et al. 2014).
BOYLE et al. (2017) have taken this principle to the extreme, combined it with further genomic and phenotype data and concluded that basically every gene expressed at the relevant time of development could be involved in a given phenotype such as human height (Fig. 4). They have called this the “omnigenic” model of quantitative traits, which claims that the sum of the influence of small effect genes (mostly regulatory ones) can be larger than the sum of the effects of genes in core pathways. Hence, this
finally suggests that FISHER’S infinitesimal model may not just be an abstract assumption, but could have a true biologic meaning.
But the data show also that there are still only a few alleles with large effects and a tail of many alleles with increasingly smaller effects (BOYLE et al. 2017) (Fig. 4). Interestingly, however, alleles with large effects tend to be rare, which has raised the possibility that they may actually be deleterious alleles that would be lost over time due to negative selection.
This brings us to the second reason for the “missing heritability” problem, namely the filtering out of low frequency alleles from standard datasets. Again, because of technical considerations (mostly related to the probability of linkage with causal variants and statistical power), only those alleles that segregate with frequencies of more than 5 % in a population (i.e. common variants) are included in standard GWAS analysis. As a result, rare alleles which constitute the vast majority of human genetic variation, and could have an impo tant contribution to overall phenotypic variation, are being overlooked. Using direct experimental approaches in yeast, it has now been shown that this assumption is indeed likely to be true and could therefore explain some of the missing heritability (FOURNIER et al. 2019, PALLARES 2019) and show that rare alleles with large phenotypic effects are likely to be recent mutations in a population (BLOOM et al. 2019).
Both, the omnigenic model, as well as the explanation for the missing heritability are still under discussion, and it is to be expected that new insights will emerge over time. An additional complication that has emerged in all GWAS is that associations may be created by indirect effects, namely non-random structure in the populations and/or hidden genetic relationships. The linkage disequilibrium effects measured through GWAS have been demonstrated to be very sensitive to population structure and this obscures also signals of selection on phenotypes (BARTON et al. 2019, BERG et al. 2019, SOHAIL et al. 2019). While the current statistical analysis pipelines try to take care of this, the problem still needs more attention in the future, especially in the context of the omnigenic model (WRAY et al. 2018, BARTON et al. 2019).
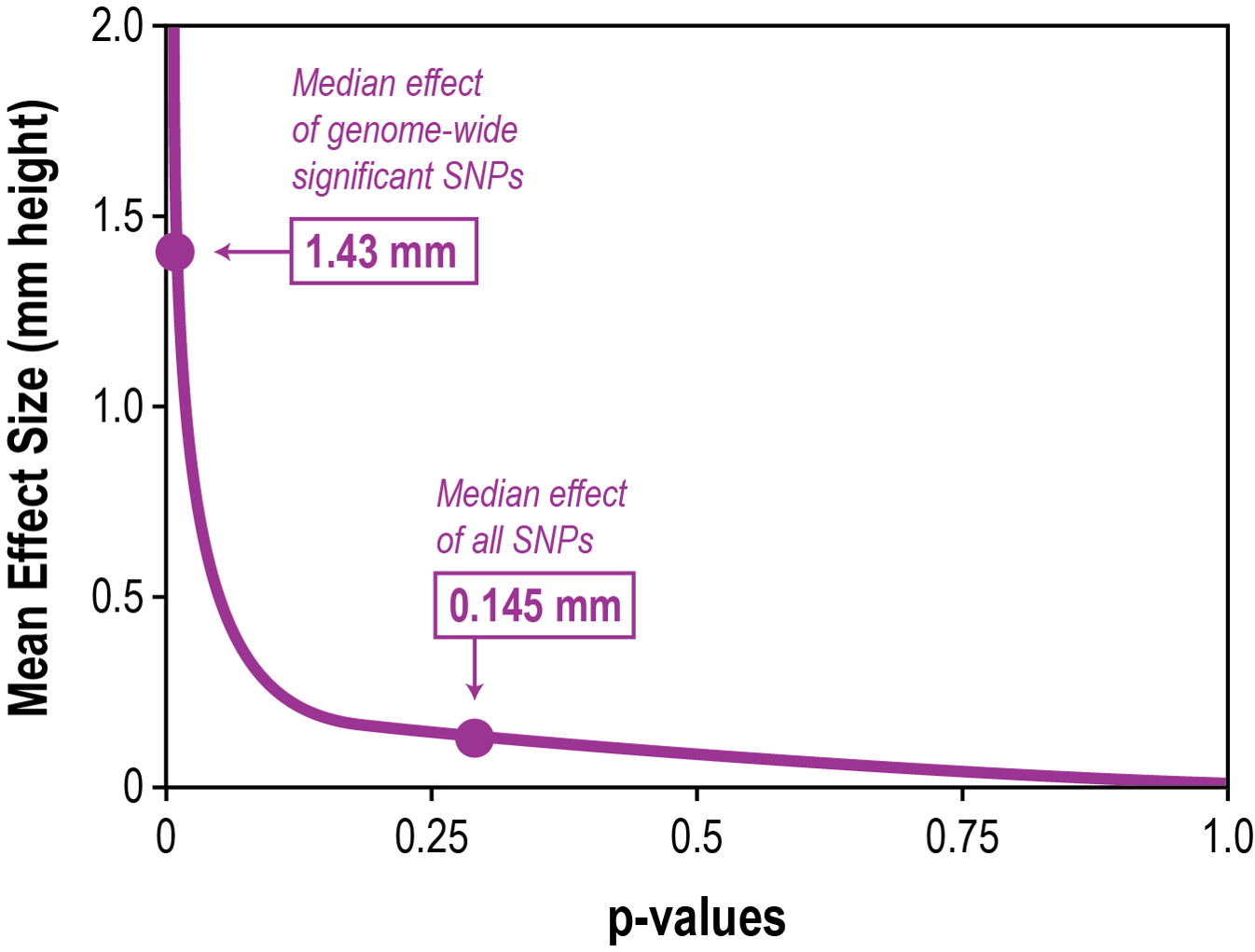
Fig. 4 Distribution of SNP effect sizes for human height data. The graph shows that there are genetic variants (SNPs) that have
strong effect on human height and these receive very high statistical support, such that they are considered as significant. The majority of genetic variants would not be considered to be significant, but can still be calculated to have an effect on size. Since these low effect SNPs are much more abundant than the large ones, they determine in total more of the final height of an individual. Figure redrawn from (BOYLE et al. 2017).
Genomic Selection
Scientists involved in livestock breeding were probably the first to accept the idea of a highly polygenic basis for quantitative traits, especially those traits relevant for commercial exploitation. However, until about 20 years ago, breeders would consider up to se eral hundred genes already as highly polygenic. Now it is commonly accepted that a large proportion of genes in the genome (in the order of 10,000) may contribute to a given trait. However, the individual effect sizes of these genes are too small to pinpoint them unequivocally and to use them for marker assisted breeding programs. This insight led to a radically new proposal for pr dicting breeding values of individuals. When most genes in the genomes contribute to a trait, then one could simply use dense marker information across the whole genome to predict the genetic value of an individual for the trait in question (MEUWISSEN et al. 2001). In practice, one trains a model based on a set of animals that had been phenotyped before, along with their associated genetic data. With this information, one can then make predictions for a new set of non-phenotyped individuals of the same breeding stock, simply by genotyping them for the same markers. In the case of dairy cow production, this approach leads to tremendous time savings, since the breeding values for bulls in terms of milk production could so far be assessed only from their daughters, i.e. with at least one generation delay (GARCIA-RUIZ et al. 2016). The impact on genetic gain was directly measurable after genomic selection regimes were implemented into the dairy cow breeding programs (Fig. 3B). This general principle was found to be widely useful in breeding programs, both for animals and plants (MEUWISSEN et al. 2016, CROSSA et al. 2017). Hence, accepting the implications for a highly polygenic nature of quantitative traits has led to a revolution in breeding procedures, the results of which are currently coming to the markets.
Evidently, the technology for high-density marker screening that is at the base of genomic selection procedures, was developed as part of the human genome project. That it would have its most immediate consequences in livestock breeding was not predictable. In the coming years, genome selection procedures will also profit from the developments in artificial intelligence, as well as automated phenotyping, since the training part is crucial to make the best predictions of breeding values (KOLTES et al. 2019).
Medical Studies versus Basic Biology Studies
It is necessary to make a distinction between medically motivated GWAS that aim to unravel disease risk phenotypes and GWAS aiming to understand basic principles of biology, like the genetic basis of variation in non-disease phenotypes. Diseases with a genetic basis are caused by deleterious mutations, or by variants that have come into mismatch with their environments. But when studying naturally occurring variation in non-disease phenotypes, one is primarily interested in genetic variants that have passed
through an adaptive phase and that are subject to stabilizing selection. This makes not only a difference for testable expectations, but also a difference for choosing cohorts, experimental designs and interpretation of the data.
In medically motivated studies it has become the norm that any particular GWAS finding should be replicated in a separate cohort to ensure that it is not a statistical artefact (CHANOCK et al. 2007). While this is a well justified cautionary procedure, it does not take into account that a given allele may have different effects in different genetic backgrounds, i.e. one is most likely filtering out possibly interesting associations when the control cohort is genetically different. Basic sciences should be particularly interested in such effects to better understand the genetic architecture as a whole, i.e. the replicability in a different cohort cannot be used as a required standard.
Another problem is that the financing of studies looking at natural phenotypes is much more limited, which has also constrained the technical and logistical means of studying such traits. This resulted in an odd situation where only the studies that found major effect loci could be published in prominent journals, leading to the impression that for natural variation one should strive to find major loci and ignore the influence of minor loci. However, such expectations might not at all be aligned with evolutionary realistic scenarios (ROCKMAN 2012). It is now rather clear that most natural phenotypes have a polygenic underpinning and that major effect loci are exceptions in special circumstances. Questions have now arisen in how far results from model organisms can help studies in humans. VISSCHER (2016, p. 378) has stated in a comment on the future of human complex trait genetics:
“I would also argue that model organisms have been largely unsuccessful in modeling complex traits in general, whether for proposed applications in human health or for potential applications in plant and animal breeding.”
One can indeed take such a view. However, the work on model organisms was mostly done within the Mendelian genetic paradigm, and it has unraveled all the mechanisms of basic biology on which also human genetics is based. Hence, the general value of studies in model organisms is undisputed in this respect. But this work has indeed yielded only few new insights into complex traits. Actually, when the function of genes and alleles is highly background dependent, one should not even expect that complex
trait results obtained in model organisms can be easily transferred to humans. Still, associations with major biochemical pathways should be reasonably conserved. For example, work on genes determining body size in mice has revealed an association with a growth control pathway (mTOR) (CHAN et al. 2012), which was then also found to be a major pathway contributing to size in humans (WOOD et al. 2014). However, one has to expect that different genes of the core pathways will be detected through the GWAS approach, since it is based on the natural variants that segregate in a given population at a given time. Hence, a gene that could be detected as a key gene through knockout genetics may be missed in a GWAS simply because it does not have differentfunctional alleles in the given test population. But since we are only at the very beginning of making connections between the two fields, it will be necessary to explore new ideas and approaches to link knowledge from model organisms to human complex trait genetics questions (LEHNER 2013).
The Genotype-Phenotype Map
The question of how the information in the genome is used to create a three-dimensional living organism (i.e. its natural phenotype) remains frustratingly poorly understood. While we have a good insight into the earliest processes of development where stem cells are formed and germ layers as well as organs are established, there is very little understanding of how development proceeds from then onwards, especially with respect to integrating and connecting organs and to establish the shape of the individual. In view of the polygenic genetics of such traits, it is actually a formidably complex question to solve. For example, the building of the skull of vertebrates requires an end product of high precision, where the senses (eyes, ears, nose) are properly i tegrated into functional units and where the feeding apparatus is optimized to deal most efficiently with the available food. At the same time, many genetic variants segregate in any population that modify the skull shape, but keep it functional in most combinations of alleles. Furthermore, the skull is also a major target of adaptive evolution, i.e. it can very easily be evolutionarily modified to respond to new food sources, new requirements for sensory reception or sexually selected modifications. The polygenic model suggests now that all these phenotypic aspects should be dependent on a large overlapping set of genes and alleles. How do they achieve the necessary integration, while re- maining evolutionarily highly flexible? If anything, these insights make the mechanisms for establishing the genotype-phenotype map even more mysterious than before. This remains therefore one of the largest
challenges in biology. It is equally a challenge for geneticists, as well as for developmental and evolutionary biologists.
In evolutionary biology the currently largest problem is to understand how adaptive selection works to shape the phenotype. Although FISHER has already provided a comprehensive treatment of this question in his 1930 book, the question of whether single loci are the main drivers of adaptation, or whether the infinitesimal model is a more appropriate model was left open for discussion. Given the technical limitations to study polygenic evolution, most current evolutionary studies and selection models on adaptation are based on single or few loci. Shaping them into new statistical tools for studying polygenic selection has only recently been taken on again (BARTON et al. 2017, JAIN and STEPHAN 2017).
Outlook
For more than 100 years, the two different views on genetics have been developed with very little experi- mental overlap. So, what are the chances to understand polygenic genetics in Mendelian mechanistic terms? GWAS have now at least provided candidate loci and are getting increasingly closer to identify candidate mutations that contribute to phenotypes. These can then theoretically serve to test them one by one with classic experimental paradigms. But is this realistic for 1000+ loci, of which each contributes only a 1000th to the phenotype – and is dependent on environmental conditions?
An experimental approach towards polygenic genetics are selection experiments. These are started from a population of individuals that all carry different natural alleles from which a phenotype of interest is selected for multiple generations. Almost any quantitative trait can be changed by continuously selecting a subset of animals from a phenotypic range distribution as parents for the next generations. The frequency of alleles involved in shaping the phenotype are expected to change in a continuous direction
with every generation under selection. By comparing the start frequency of alleles with the end frequencies, one has the prospect to identify the genes that have contributed to the selected phenotype. However, allele frequency changes can also occur by random drift, especially since selection entails also some form of genetic bottleneck. To better distinguish between such random effects and the directed effects, one can set up parallel lines of selection as replicates. One can then expect that the random effects cancel each other out in the parallels, while the directed effects remain.
Although the possible power of parallel selection has long been realized (FALCONER 1973), the need for high density genotyping to evaluate all genetic variation has impeded its application. This has changed in the past decade with the increasing availability of high-density SNP arrays and nowadays with the option to use whole genome sequencing data for genotyping. Proof of principle experiments have emerged that confirmed the power of parallel selection to map complex traits (CHAN et al. 2012, LONG et al. 2015, HUANG et al. 2018, BARGHI et al. 2019, CASTRO et al. 2019). Hence, rather than just describing genetic associations with phenotypes, one has a tool that is based on experimentally manipulating a phenotype in a polygenic context.
However, all of these approaches still rely on naturally segregating alleles, which were shaped by evolutionary processes. One of the powers of the classic genetic approach is the generation of random unbiased mutants for phenotypic analysis. It may therefore be profitable to combine association studies with mutagenesis experiments. For example, if the base population for an association study is derived from a mutagenesis experiment of an inbred strain, rather than from wildtype variation, one has the possibility to link random unbiased mutations to a phenotype of interest. By doing this in independent mutagenesis experiments, one should be able to unravel the components of the network that contribute to the phenotype in a systematic way. This may eventually provide a bridge between the genome wide natural variation examined in GWAS and the more limited synthetic variation that forms the basis of
Mendelian studies.
Acknowledgements
We thank Derek CAETANO-ANOLLES for drawing the figures in a consistent design.
References
Statement on Literature Search and Citations
The topic is rather broad, both with respect to covering an historical time frame, as well as the subtopics that are addressed. An exhaustive literature search would turn up thousands of highly relevant papers. It was therefore necessary to focus the citations on key papers. On the one hand we focused on highly cited books and reviews covering the subtopics. But we cite also some recent key papers that we consider to establish new principles, even if their citation rate is not yet high.
ALLEN, H. L., ESTRADA, K., LETTRE, G., BERNDT, S. I., WEEDON, M. N., RIVADENEIRA, F., WILLER, C. J., JACKSON, A. U., VEDANTAM, S., RAYCHAUDHURI, S., et al.: Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838 (2010). doi:10.1038/nature09410
BARGHI, N., TOBLER, R., NOLTE, V., JAKSIC, A. M., MALLARD, F., OTTE, K. A., DOLEZAL, M., TAUS, T., KOFLER, R., and SCHLÖTTERER, C.: Genetic redundancy fuels polygenic adaptation in Drosophila. Plos Biology 17 (2019).
doi:10.1371/journal.pbio.3000128
BARNETT, A. G., VAN DER POLS, J. C., and DOBSON, A. J.: Regression to the mean: what it is and how to deal with it. International Journal of Epidemiology 34, 215–220 (2005). doi:10.1093/ije/dyh299
BARTON, N., HERMISSON, J., and NORDBORG, M.: Population genetics: Why structure matters. Elife 8 (2019). doi:10.7554/eLife.45380
BARTON, N. H., ETHERIDGE, A. M., and VEBER, A.: The infinitesimal model: Definition, derivation, and implications. Theoretical Population Biology 118, 50–73 (2017). doi:10.1016/j.tpb.2017.06.001
BERG, J. J., HARPAK, A., SINNOTT-ARMSTRONG, N., JOERGENSEN, A. M., MOSTAFAVI, H., FIELD, Y., BOYLE, E. A., ZHANG, X. J., RACIMO, F., PRITCHARD, J. K., , and COOP, G.: Reduced signal for polygenic adaptation of height in UK Biobank. Elife 8 (2019). doi:10.7554/eLife.39725
BLOOM, J. S., BOOCOCK, J., TREUSCH, S., SADHU, M. J., DAY, L., OATES-BARKER, H., and KRUGLYAK, L.: Rare variants contribute disproportionately to quantitative trait variation in yeast. Elife 8 (2019). doi:10.7554/eLife.49212
BOYLE, E. A., LI, Y. I., and PRITCHARD, J. K.: An expanded view of complex traits: From polygenic to omnigenic. Cell 169, 1177–1186 (2017). doi:10.1016/j.cell.2017.05.038
BURTON, P. R., CLAYTON, D. G., CARDON, L. R., CRADDOCK, N., DELOUKAS, P., DUNCANSON, A., KWIATKOWSKI, D. P., MCCARTHY, M. I., OUWEHAND, W. H., SAMANI, N. J., et al.: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007). doi:10.1038/nature05911
CASTRO, J. P. L., YANCOSKIE, M. N., MARCHINI, M., BELOHLAVY, S., HIRAMATSU, L., KUCKA, M., BELUCH, W. H., NAUMANN, R., SKUPLIK, I., COBB, J., BARTON, N. H., ROLIAN, C., and CHAN, Y. F..: An integrative genomic analysis of the Longshanks selection experiment for longer limbs in mice. Elife 8 (2019). doi:10.7554/eLife.42014
CHAN, Y. F., JONES, F. C., MCCONNELL, E., BRYK, J., BUENGER, L., and TAUTZ, D.: Parallel selection mapping using artificially selected mice reveals body weight control loci. Current Biology 22, 794–800 (2012). doi:10.1016/j.cub.2012.03.011
CHANDLER, C. H., CHARI, S., and DWORKIN, I.: Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution. Trends in Genetics 29, 358–366 (2013). doi:10.1016/j.tig.2013.01.009
CHANOCK, S. J., MANOLIO, T., BOEHNKE, M., BOERWINKLE, E., HUNTER, D. J., THOMAS, G., HIRSCHHORN, J. N., ABECASIS, G., ALTSHULER, D., BAILEY-WILSON, J. E., BROOKS, L. D., CARDON, L. R., DALY, M., DONNELLY, P., FRAUMENI Jr., J. F.,
FREIMER, N. B., GERHARD, D. S., GUNTER, C., GUTTMACHER, A. E., GUYER, M. S., HARRIS, E. L., HOH, J., HOOVER, R., KONG, C. A., MERIKANGAS, K. R., MORTON, C. C., PALMER, L. J., PHIMISTER, E. G., RICE, J. P., ROBERTS, J., ROTIMI, C., TUCKER, M. A., VOGAN,K. J., WACHOLDER, S., WIJSMAN, E. M., WINN, D. M., and COLLINS, F. S.: Replicating genotype-phenotype associations. Nature 447, 655–660 (2007). doi:10.1038/447655a
Crossa, J., Pérez-Rodríguez, P., Cuevas, J., Montesinos-López, O., Jarquín, D., De Los Campos, G., BURGUEÑO, J., GONZÁLEZ-CAMACHO, J. M., PÉREZ-ELIZALDE, S., BEYENE, Y., DREISIGACKER, S., SINGH, R., ZHANG, X., GOWDA, M., ROORKIWAL, M., RUTKOSKI, J., AND VARSHNEY, R. K.: Genomic selection in plant breeding: Methods, models, and perspectives. Trends in Plant Science 22, 961–975 (2017). doi:10.1016/j.tplants.2017.08.011
DARWIN, C.: On the Origin of Species by Means of Natural Selection. Or the Preservation of Favored Races in the Struggle for Life. London: Murray 1859 EICHLER, E. E., FLINT, J., GIBSON, G., KONG, A., LEAL, S. M., MOORE, J. H., and NADEAU, J. H.: VIEWPOINT. Missing heritability and strategies for finding the underlying causes of complex disease. Nature Reviews Genetics 11, 446–450 (2010). doi:10.1038/nrg2809
EINSTEIN, A., and INFELD, L.: The Evolution of Physics: The Growth of Ideas from Early Concepts to Relativity and Quanta. Cambridge: Cambridge University Press 1938
FALCONER, D. S.: Replicated selection for body weight in mice. Genetical Research 22, 291–321 (1973). doi:10.1017/s0016672300013094
FISHER, R. A.: The correlation between relatives on the supposition of Mendelian inheritance. Transactions of the Royal Society of Edinburgh 52, 399–433 (1918)
FISHER, R. A.: The Genetical Theory of Natural Selection. Oxford: Clarendon Press 1930
FOURNIER, T., ABOU SAADA, O., HOU, J., PETER, J., CAUDAL, E., and SCHACHERER, J.: Extensive impact of low-frequency variants on the phenotypic landscape at population-scale. Elife 8 (2019). doi:10.7554/eLife.49258
GALTON, F.: Natural Inheritance. London: Macmillan 1889
GALTON, F.: Eugenics: Its definition, scope, and aims. American Journal of Sociology 10, 1–25 (1904). doi:10.1086/211280
GANNA, A., VERWEIJ, K. J. H., NIVARD, M. G., MAIER, R., WEDOW, R., BUSCH, A. S., ABDELLAOUI, A., GUO, S., SATHIRAPONGSASUTI, J. F., LICHTENSTEIN, P., LUNDSTRÖM, S., LÅNGSTRÖM, N., AUTON, A., HARRIS, K. M., BEECHAM, G. W., MARTIN, E. R., SANDERS, A. R., PERRY, J. R. B., NEALE, B. M., and ZIETSCH, B. P.: Large-scale GWAS reveals insights into the genetic architecture of same-sex sexual behavior. Science 365, 882–+ (2019). doi:10.1126/science.aat7693
GARCIA-RUIZ, A., Cole, J. B., VANRADEN, P. M., WIGGANS, G. R., RUIZ-LOPEZ, F. J., and VAN TASSELL, C. P.: Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proceedings of the National Academy of Sciences USA 113, E3995–E4004 (2016). doi:10.1073/pnas.1519061113
HERNSTEIN, R., and MURRAY C.: The Bell Curve: Intelligence and Class Structure in American Life. New York (NY,UuSA): Free Press 1994
HUANG, J., LI, J., ZHOU, J., WANG, L., YANG, S. H., HURST, L. D., LI, W. H., and TIAN, D. C.: Identifying a large number of high-yield genes in rice by pedigree analysis, whole-genome sequencing, and CRISPR-Cas9 gene knockout. Proceedings of the National Academy of Sciences USA 115, E7559–E7567 (2018). doi:10.1073/pnas.1806110115
JAIN, K., and STEPHAN, W.: Modes of rapid polygenic adaptation. Molecular Biology and Evolution 34, 3169–3175 (2017). doi:10.1093/molbev/msx240
JOEL, D., GARCIA-FALGUERAS, A., and SWAAB, D.: The complex relationships between sex and the brain. Neuroscientist 26, 156–169 (2020). doi:10.1177/1073858419867298
KOLTES, J. E., COLE, J. B., CLEMMENS, R., DILGER, R. N., KRAMER, L. M., LUNNEY, J. K., MCCUE, M. E., MCKAY, S. D., MATEESCU, R. G., MURDOCH, B. M., REUTER, R., REXROAD, C. E., ROSA, G. J. M., SERÃO, N. L., WHITE, S. N., WOODWARD-GREENE, M. J., WORKU, M., ZHANG, H., and REECY, J. M.: A vision for development and utilization of high-throughput phenotyping and big data analytics in livestock. Frontiers in Genetics 10 (2019). doi:10.3389/fgene.2019.01197
LEHNER, B.: Genotype to phenotype: lessons from model organisms for human genetics. Nature Reviews Genetics 14, 168–178 (2013). doi:10.1038/nrg3404
LONG, A., LITI, G., LUPTAK, A., and TENAILLON, O.: Elucidating the molecular architecture of adaptation via evolve and resequence experiments. Nature Reviews Genetics 16, 567–582 (2015). doi:10.1038/nrg3937
MANEY, D. L.: Perils and pitfalls of reporting sex differences. Philosophical Transactions of the Royal Society B-Biological Sciences 371 (2016). doi:10.1098/rstb.2015.0119
MANOLIO, T. A., COLLINS, F. S., COX, N. J., GOLDSTEIN, D. B., HINDORFF, L. A., HUNTER, D. J., MCCARTHY, M. I., RAMOS, E. M., CARDON, L. R., CHAKRAVARTI, A., CHO, J. H., GUTTMACHER, A. E., KONG, A., KRUGLYAK, L., MARDIS, E., ROTIMI, C. N., SLATKIN, M., VALLE, D., WHITTEMORE, A. S., BOEHNKE, M., CLARK, A. G., EICHLER, E. E., GIBSON, G., HAINES, J. L., MACKAY, T. F. C., MCCARROLL, S. A., and VISSCHER, P. M.: Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009). doi:10.1038/nature08494
MENDEL, G.: Versuche über Pflanzen-Hybriden. Verhandlungen des naturforschenden Vereines in Brünn IV, 3–47 (1866)
MEUWISSEN, T. H. E., HAYES, B. J., and GODDARD, M. E.: Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829 (2001). pmid:11290733 pmcid:pmc1461589
MEUWISSEN, T., HAYES, B., and GODDARD, M.: Genomic selection: A paradigm shift in animal breeding. Animal Frontiers 6, 6–14 (2016). doi:10.2527/af.2016-0002
NARASIMHAN, V. M., HUNT, K. A., MASON, D., BAKER, C. L., KARCZEWSKI, K. J., BARNES, M. E. R., BARNETT, A. H., BATES, C., BELLARY, S., BOCKETT, N. A., GIORDA, K., GRIFFITHS, C. J., HEMINGWAY, H., JIA, Z., KELLY, M. A., KHAWAJA, H. A., LEK, M., MCCARTHY, S., MCEACHAN, R., OʼDONNELL-LURIA, A., PAIGEN, K., PARISINOS, C. A., SHERIDAN, E., SOUTHGATE, L., TEE, L., THOMAS, M., XUE, Y., SCHNALL-LEVIN, M., PETKOV, P. M., TYLER-SMITH, C., MAHER, E. R., TREMBATH, R. C., MACARTHUR, D. G., WRIGHT, J., DURBIN, R., and VAN HEEL, D. A.: Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477 (2016). doi:10.1126/science.aac8624
NOLAN, P. M., PETERS, J., STRIVENS, M., ROGERS, D., HAGAN, J., SPURR, N., GRAY, I. C., VIZOR, L., BROOKER, D., WHITEHILL, E., WASHBOURNE, R., HOUGH, T., GREENAWAY, S., HEWITT, M., LIU, X., MCCORMACK, S., PICKFORD, K., SELLEY, R., WELLS, C., TYMOWSKA-LALANNE, Z., ROBY, P., GLENISTER, P., THORNTON, C., THAUNG, C., STEVENSON, J. A., ARKELL, R., MBURU, P., HARDISTY, R., KIERNAN, A., ERVEN, A., STEEL, K. P., VOEGELING, S., GUENET, J. L., NICKOLS, C., SADRI, R., NAASE, M., ISAACS, A., DAVIES, K., BROWNE, M., FISHER, E. M., MARTIN, J., RASTAN, S., BROWN, S. D. M., and HUNTER, J.: A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nature Genetics 25, 440–443 (2000). doi:10.1038/78140
NUESSLEIN-VOLHARD, C., and WIESCHAUS, E.: Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801 (1980). doi:10.1038/287795a0
PALLARES, L. F.: Genetic Variation: Searching for solutions to the missing heritability problem. Elife 8 (2019). doi:10.7554/eLife.53018
RANCATI, G., MOFFAT, J., TYPAS, A., and PAVELKA, N.: Emerging and evolving concepts in gene essentiality. Nature Reviews Genetics 19, 34–49 (2018). doi:10.1038/nrg.2017.74
REIS, H. T., and CAROTHERS, B. J.: Black and white or shades of gray: Are gender differences categorical or dimensional? Current Directions in Psychological Science 23, 19–26 (2014). doi:10.1177/0963721413504105
ROCKMAN, M. V.: The QTN program and the alleles that matter for evolution: all that´s gold does not glitter. Evolution 66, 1–17 (2012). doi:10.1111/j.1558-5646.2011.01486.x
SITTIG, L. J., CARBONETTO, P., ENGEL, K. A., KRAUSS, K. S., BARRIOS-CAMACHO, C. M., and PALMER, A. A.: Genetic background limits generalizability of genotype-phenotype relationships. Neuron 91, 1253–1259 (2016). doi:10.1016/j.neuron.2016.08.013
SOHAIL, M., MAIER, R. M., GANNA, A., BLOEMENDAL, A., MARTIN, A. R., Turchin, M. C., CHIANG, C. W. K., HIRSCHHORN, J., DALY, M. J., PATTERSON, N., NEALE, B., MATHIESON, I., REICH, D., and SUNYAEV, S. R.: Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8 (2019). doi:10.7554/eLife.39702
STANLEY, H.: Mr. Galton on natural inheritance. Nature 40, 642–643 (1889). doi:10.1038/040642c0
THODAY, J. M., and THOMPSON, J. N.: The number of segregating genes implied by continuous variation. Genetica 46, 335–344 (1976). doi:10.1007/BF00055476
VISSCHER, P. M.: Human complex trait genetics in the 21st century. Genetics 202, 377–379 (2016). doi:10.1534/genetics.115.180513
VISSCHER, P. M., BROWN, M. A., MCCARTHY, M. I., and YANG, J.: Five years of GWAS discovery. American Journal of Human Genetics 90, 7–24 (2012). doi:10.1016/j.ajhg.2011.11.029
VISSCHER, P. M., HEMANI, G., VINKHUYZEN, A. A. E., CHEN, G. B., LEE, S. H., WRAY, N. R., GODDARD, M. E., and YANG, J.: Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. Plos Genetics 10 (2014). doi:10.1371/journal.pgen.1004269
WEEDEN, N. F.: Are Mendelʼs data reliable? The perspective of a pea geneticist. Journal of Heredity 107, 635–646 (2016). doi:10.1093/jhered/esw058
WHITE, D., and RABAGO-SMITH M.: Genotype-phenotype associations and human eye color. Journal of Human Genetics 56, 5–7 (2011). doi:10.1038/jhg.2010.126
WOOD, A. R., ESKO, T., YANG, J., VEDANTAM, S., PERS, T. H., GUSTAFSSON, S., CHUN, A. Y., ESTRADA, K., LUAN, J., KUTALIK, Z., et al.: Defining the role of common variation in the genomic and biological architecture of adult human height. Nature Genetics 46, 1173–1186 (2014). doi:10.1038/ng.3097
WRAY, N. R., WIJMENGA, C., SULLIVAN, P. F., YANG, J., and VISSCHER, P. M.: Common disease is more complex than implied by the core gene omnigenic model. Cell 173, 1573–1580 (2018). doi:10.1016/j.cell.2018.05.051
YANG, J. A., BENYAMIN, B., MCEVOY, B. P., GORDON, S., HENDERS, A. K., NYHOLT, D. R., MADDEN, P. A., HEATH, A. C., MARTIN, N. G., MONTGOMERY, G. W., GODDARD, M. E., and VISSCHER, P. M.: Common SNPs explain a large proportion of the heritability for human height. Nature Genetics 42, 565–U131 (2010). doi:10.1038/ng.608
YANG, J., MANOLIO, T. A., PASQUALE, L. R., BOERWINKLE, E., CAPORASO, N., CUNNINGHAM, J. M., ANDRADE, M. DE, FEENSTRA, B., FEINGOLD, E., HAYES, M. G., HILL, W. G., LANDI, M. T., ALONSO, A., LETTRE, G., LIN, P., LING, H., LOWE,
W., MATHIAS, R. A., MELBYE, M., PUGH, E., CORNELIS, M. C., WEIR, B. S., GODDARD, M. E., and VISSCHER, P. M.: Genome partitioning of genetic variation for complex traits using common SNPs. Nature Genetics 43, 519–U544 (2011). doi:10.1038/ng.823